Introduzione
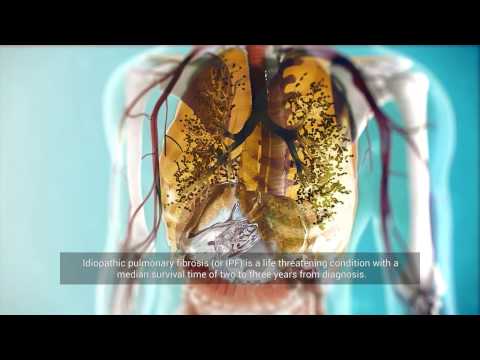
La fibrosi polmonare è una malattia in cui vi è una formazione patologica di tessuto cicatriziale nei polmoni in modo che non possano svolgere completamente la loro funzione per fornire ossigeno al corpo. Questa condizione peggiora significativamente la qualità della vita del paziente. In alcuni casi, la progressione della malattia può portare a insufficienza polmonare e / o cardiaca e morte. I farmaci medici moderni non possono invertire i cambiamenti patologici nella fibrosi polmonare. Metodi alternativi di trattamento della patologia possono includere le cellule staminali, poiché le loro proprietà studiate implicano la capacità di fermare il processo infiammatorio, nonché di stimolare il recupero dei tessuti danneggiati.
Informazioni sulla fibrosi polmonare
La fibrosi polmonare è associata a cicatrici del tessuto polmonare a causa della sua degenerazione fibrotica. La crescita del tessuto cicatriziale, causata di regola dall’infiammazione, porta alla compressione degli alveoli, che non possono più svolgere pienamente la loro funzione, ovveroi garantire lo scambio di gas nel corpo. Gli alveoli sono le vescicole polmonari in cui l’aria inalata entra in contatto con il sangue. Le lesioni fibrotiche possono interessare una parte del polmone o l’intero organo, in uno o entrambi i polmoni.

La fibrosi polmonare, o pneumofibrosi, è una malattia progressiva con cambiamenti irreversibili nel tessuto polmonare, le cui complicanze e conseguenze includono:
- Insufficienza respiratoria cronica (ossigeno insufficiente per il corpo).
- Ipertensione polmonare (pressione sanguigna alta nelle arterie polmonari).
- Cuore polmonare cronico e altre malattie cardiovascolari.
- Cancro ai polmoni.
- Infezione respiratoria secondaria (compreso lo sviluppo di polmonite ad alto rischio di morte).
Le cause della fibrosi polmonare
Non è sempre possibile determinare il motivo esatto che causa la cicatrizzazione polmonare. La crescita patologica del tessuto connettivo nei polmoni può essere provocata da allergie, infezioni polmonari, fattori genetici, radiazioni, traumi e altri processi e circostanze.
Quando è impossibile determinare la causa esatta della patologia, si parla di fibrosi polmonare idiopatica (IPF). In una tale malattia, una combinazione di esposizione ambientale, fattori genetici e altri fattori non identificati è considerata un provocatore.
Ci sono anche prove che il fumo (sia nel presente che nel passato), oltre a una storia familiare gravata (fino al 20% dei pazienti), gioca un ruolo fondamentale. Quando almeno due o più membri della famiglia biologica primaria (genitore, fratello o figlio) hanno avuto manifestazioni cliniche di IPF, la malattia è classificata come polmonite interstiziale familiare (FIP). I parenti di primo grado dei pazienti con FIP hanno un rischio notevolmente aumentato di sviluppare malattia polmonare interstiziale (ILD).
Fibrosi polmonare interstiziale
La malattia polmonare interstiziale (MPI), nota anche come malattia polmonare parenchimale diffusa (MPPD), è un gruppo di patologie, inclusa la fibrosi polmonare, in cui il tessuto interstiziale (tessuto connettivo del polmone) è danneggiato. L’MPI è associata a infiammazione e cicatrizzazione del tessuto polmonare.
Il tessuto interstiziale è il tessuto connettivo dei polmoni che funge da spina dorsale per gli alveoli e i vasi sanguigni. Nella fibrosi, a causa dell’eccessiva crescita del tessuto connettivo nel polmone, gli alveoli vengono compressi e successivamente sostituiti con tessuto connettivo. Di conseguenza, lo scambio di gas negli alveoli diventa difficile, con conseguente ipossia: il corpo inizia a soffrire di mancanza di ossigeno.
A differenza della fibrosi polmonare idiopatica (FPI), la causa dell’MPI può essere determinata in modo affidabile. Questi motivi includono:
- lesioni infettive
- malattie autoimmuni / del tessuto connettivo
- predisposizione genetica
- tumori maligni
- sostanze nocive inalate (vapori di mercurio, polvere minerale, ecc.)
- alcuni farmaci (ad esempio, alcuni farmaci antiaritmici, nitrofurantoina e altri)
Contattare
Ottieni una consulenza online gratuita per conoscere i risultati attesi della terapia con cellule staminali >>>

Medical Advisor, Swiss Medica doctor
La pandemia di coronavirus ha aumentato il numero dei casi di fibrosi polmonare. Ora è noto che la cicatrizzazione polmonare è una delle conseguenze più frequenti e più gravi dell’esposizione virale sul corpo umano. Il meccanismo di sviluppo della fibrosi polmonare causata da COVID-19 è associato a una risposta immunitaria errata, che porta al danneggiamento delle cellule epiteliali polmonari e delle cellule endoteliali microvascolari, specialmente in combinazione con la ventilazione polmonare artificiale.

Inoltre, secondo i ricercatori e i dati statistici, diventa chiaro che i pazienti con lieve infezione da COVID-19, anche dopo il miglioramento e la dimissione dall’ospedale, hanno un potenziale rischio nascosto di sviluppare fibrosi polmonare.
Per prevenire la progressione della fibrosi polmonare, indotta dal coronavirus, è fondamentale iniziare il trattamento di questa complicanza il prima possibile.
Segni e sintomi della fibrosi polmonare
Il decorso clinico della fibrosi polmonare è associato a una diminuzione del volume polmonare, che costringe l’organismo ad adattarsi a questa conseguenza negativa. I cambiamenti compensatori si manifestano come mancanza di respiro, che è il sintomo iniziale più frequente riportato dai pazienti con PIF. All’inizio, la mancanza di respiro si verifica solo durante lo sforzo fisico, ma successivamente anche a riposo.
Il paziente con PIF affronta anche sintomi come tosse secca, affaticamento e perdita di peso inspiegabile.
Inoltre, potrebbero essere osservate le caratteristiche cliniche di una malattia del tessuto connettivo sottostante (TCS), come la artralgia (dolore in diverse articolazioni) o sintomi di sicca (secchezza delle ghiandole esocrine, in particolare gli occhi e la bocca).
Il principale segnale esterno di PIF è noto come «dita bastonate», una deformità delle dita delle mani o dei piedi caratterizzata dall’allargamento delle punte delle dita. Questo sintomo è associato a livelli di ossigeno nel sangue cronicamente bassi.
Le fasi iniziali della malattia si verificano più spesso senza manifestazioni. Ad esempio, negli individui asintomatici a rischio di polmonite interstiziale familiare (PIF), è possibile identificare diagnosticamente i segni di alterazioni istologiche nel tessuto polmonare.
Diagnosi della fibrosi polmonare
Si ritiene che la fibrosi polmonare come malattia non sia sufficientemente diagnosticata. Molto spesso, viene rilevata in una fase abbastanza avanzata. Comunemente, la malattia è identificata da un’anomalia su una tomografia computerizzata ad alta risoluzione (HRCT) del torace.
Ci sono prove che la radiografia del torace è meno utile della HRCT nella valutazione dei pazienti con sospetta fibrosi.
La broncoscopia con lavaggio broncoalveolare (BLB) e la biopsia transbronchiale vengono anche utilizzate per determinare le condizioni del tessuto polmonare.

Inoltre, la diagnostica di laboratorio di vari indicatori (marcatori biologici) può fornire dati oggettivamente misurabili sui processi che si verificano nel corpo, sia normali che patologici. Nella fibrosi polmonare, questi biomarcatori aiutano a diagnosticare la malattia, rivelare la suscettibilità alla malattia e prevedere l’efficacia specifica del farmaco. Possono anche prevedere con precisione il decorso della malattia e la sua prognosi in un paziente con IPF.
Trattamenti tradizionali e moderni per MPI e fibrosi polmonare
Mentre la fibrosi polmonare può richiedere anni per svilupparsi, nella maggior parte dei pazienti la malattia viene diagnosticata nella sua fase avanzata, quando si può fare poco per influenzarne la progressione. I trattamenti esistenti mirano a mantenere le prestazioni polmonari del paziente e uno standard di vita accettabile.
Nel metodo comune di trattamento della pneumofibrosi vengono utilizzati gli ormoni della corteccia surrenale (prednisone) e, se non sono abbastanza efficaci, vengono utilizzati azatioprina e ciclofosfano. Inoltre, viene somministrato ossigeno per inalazione. Recentemente, i farmaci pirfenidone e nintedanib hanno dimostrato di rallentare la progressione dell’PIF.
Tuttavia, la fibrosi polmonare è una malattia progressiva, e nessun agente o trattamento anti fibrotico (a parte un trapianto di polmone) è noto per avere un impatto sullo scarso tasso di sopravvivenza associato a questa malattia.
Metodi alternativi per il trattamento della fibrosi polmonare e del MPI includono l’uso di prodotti a base di cellule staminali. I cambiamenti patologici che si verificano nei polmoni con fibrosi sono considerati irreversibili, a causa del fatto che il corpo ha una scarsa capacità di guarire il tessuto cicatriziale. Tuttavia, quando immettiamo cellule staminali nel corpo nella quantità di una dose terapeutica, spesso questo è sufficiente per avviare i processi di rigenerazione e rallentare il decorso della cicatrizzazione del tessuto polmonare. Queste cellule possono essere iniettate attraverso una flebo IV (per via endovenosa) o per inalazione per il rilascio mirato di cellule nell’area danneggiata.
Contattare
Contatta il nostro consulente medico per conoscere i vantaggi che puoi ottenere dalla terapia con cellule staminali >>>
Medical Advisor, Swiss Medica doctor
Terapia con le cellule staminali per la fibrosi polmonare
La terapia cellulare è nota come un moderno trattamento sperimentale che utilizza la capacità del corpo di curarsi.
Come agiscono le cellule staminali infuse nella fibrosi? La particolarità del «lavoro» delle cellule staminali nell’organismo è che rispondono ai segnali inviati dalle cellule dei tessuti danneggiati, e producono in risposta citochine (molecole proteiche che trasmettono segnali intercellulari), che stimolano la formazione delle nuove cellule richieste.
Studi su cellule staminali mesenchimali (CSM) allogeniche (donatrici) hanno dimostrato che esse si adattano ai siti di danno polmonare, e contribuiscono alla rigenerazione dei tessuti, diminuendo l’infiammazione cronica delle vie aeree e ripristinando l’equilibrio del liquido alveolare nel danno polmonare acuto.
Attualmente sono in corso ulteriori studi clinici sulle cellule staminali per la malattia polmonare.
Quali sono i possibili risultati dopo la terapia con le cellule staminali?
Utilizzando cellule staminali per infusioni endovenose nel trattamento della fibrosi polmonare, possiamo ottenere i seguenti effetti:
- Rallentamento o arresto del processo di cicatrizzazione, «calmando» il sistema immunitario del paziente (le cellule staminali hanno una capacità antinfiammatoria).
- Rigenerazione parziale del tessuto polmonare.
- Formazione di nuove reti vascolari, che contribuiscono al rinnovamento dei tessuti.
- Sollievo dai sintomi del paziente.
Secondo uno studio clinico, le infusioni endobronchiali di cellule staminali hanno aumentato il numero di indicatori per la qualità della vita. L’86% dei partecipanti allo studio, che sono stati osservati per 12 mesi, ha dimostrato uno stato funzionale e una prestazione fisica stabili.
Pertanto, il ruolo principale della terapia, basata sull’introduzione di cellule staminali, è rallentare la progressione della malattia, stabilizzare la funzione polmonare e mantenere lo stato attuale del paziente.
Guarda la video recensione dei pazienti con BPCO (in italiano) per scoprire come la terapia con cellule staminali può aiutare nella malattia polmonare.

Va tenuto presente che la quantità di miglioramento in ciascun paziente può differire, poiché dipende da diversi fattori: la negligenza della malattia, la presenza di ulteriori patologie, l’età del paziente e la «risposta» individuale al trattamento con cellule staminali.
Sicurezza delle cellule staminali nella fibrosi polmonare
Studi clinici di terapia, basati su cellule staminali adulte, hanno dimostrato un profilo di sicurezza accettabile di questo approccio. In particolare, è stato riferito che durante l’intero periodo di uno studio con 72 infusioni, non sono stati riportati effetti collaterali che potrebbero essere considerati gravi o clinicamente significativi.
È attualmente in discussione se le CSM possano differenziarsi in fibroblasti, data la loro comune origine mesodermica, e accelerare la cascata fibrosa. Tuttavia, non ci sono studi preclinici o sull’uomo che hanno dimostrato questa relazione.
Nessuno dei pazienti ha manifestato formazione di tessuto anormale, come riportato da una TAC su tutto il corpo eseguita 12 mesi dopo la prima somministrazione di cellule staminali. Lo stesso risultato positivo di sicurezza è stato ottenuto 24 mesi dopo la prima infusione.
Un altro studio dimostra che le infusioni di cellule staminali sono state ben tollerate in tutti i pazienti e non sono stati riportati eventi avversi gravi emergenti dal trattamento.
Сontattare
Contatta il nostro consulente medico per ottenere una pre-valutazione online gratuita per il tuo caso personale >>>
Medical Advisor, Swiss Medica doctor
Riferimenti bibliografici:
World Health Organisation. The top 10 causes of death. 24 May 2018.
Mesenchymal Stem Cell as Therapeutic Modality in Interstitial Pulmonary Fibrosis. Clinical Trial.